
Pharming is commercializing and developing a portfolio of innovative medicines, including small molecules and biologics.
Our Portfolio
The following chart summarizes our main product candidate portfolio.
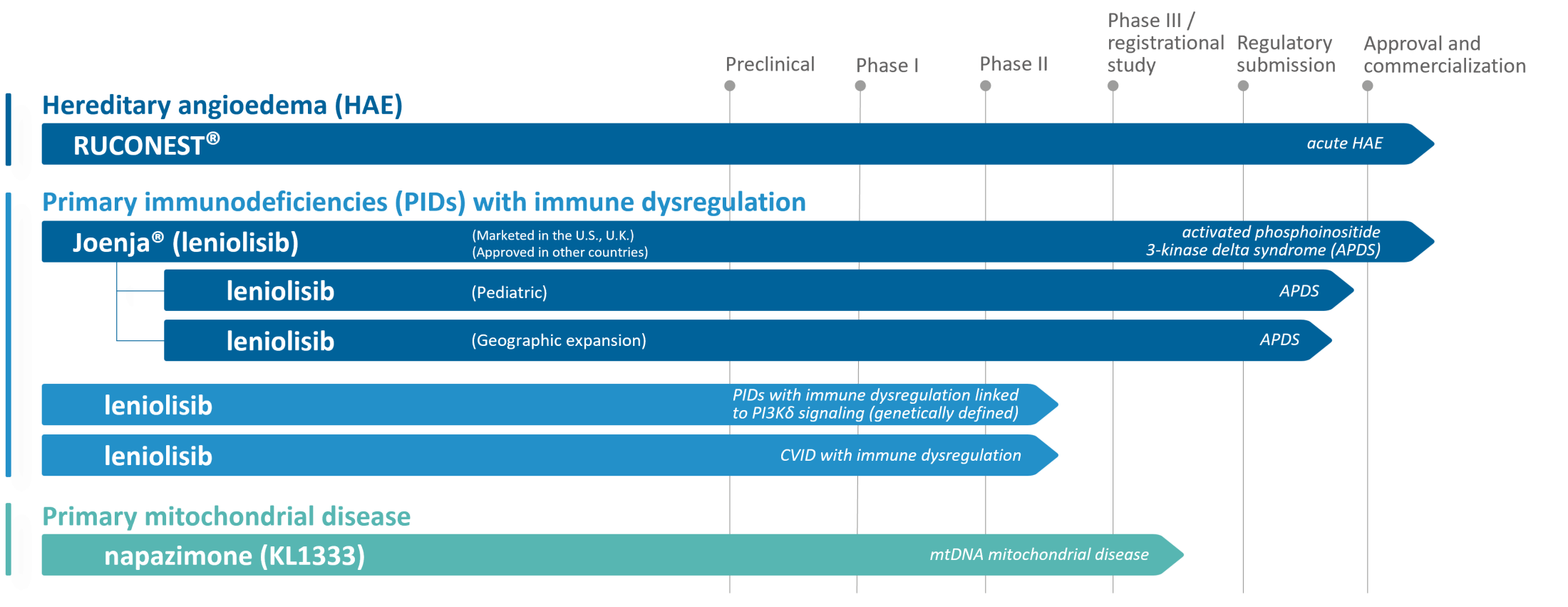
DEVELOPMENT PROGRAMS
Activated Phosphoinositide 3-kinase Delta (PI3Kδ) syndrome (APDS)
Activated Phosphoinositide 3-kinase Delta (PI3Kδ) Syndrome, or APDS is a rare, genetic, and progressive primary immunodeficiency. Discovered in 2013, APDS is a disorder that impairs the immune system and the function of the white blood cells that recognize and attack viruses and bacteria to prevent infection.
Primary immunodeficiences (PIDs) with immune dysregulation
We are developing leniolisib for additional primary immunodeficiencies, or PIDs, which present with similar clinical manifestations to APDS but which affect significantly more patients than APDS. These include (i) genetically identifiable PIDs with immune dysregulation linked to altered PI3Kδ signaling and (ii) common variable immunodeficiency, or CVID, with immune dysregulation identified independently of genetics.
(i) PIDs with immune dysregulation linked to altered PI3Kδ signaling
Primary immunodeficiencies (PIDs) with immune dysregulation linked to altered PI3Kẟ signaling in lymphocytes include ALPS-FAS, CTLA4 haploinsufficiency, NFKB1 haploinsufficiency and PTEN deficiency, among others. The unique genetic drivers in these PIDs lead to similar clinical phenotypes and unmet medical need to APDS. A Phase II proof of concept clinical trial is ongoing.
READ MORE ABOUT PIDs WITH IMMUNE DYSREGULATION
(ii) Common variable immunodeficiency (CVID) with immune dysregulation
CVID represents the largest group of symptomatic PID patients, where approximately 50% display autoimmune, lymphoproliferative and/or end-organ lympho-infiltrative clinical manifestations driven by immune dysregulation. The majority of CVID patients with immune dysregulation exhibit a spectrum of clinical manifestations with similarities to APDS. A Phase II proof of concept clinical trial is ongoing.
READ MORE ABOUT CVID WITH IMMUNE DYSREGULATION
napazimone (KL1333) for mitochondrial DNA-driven primary mitochondrial disease
Mitochondrial disease is a rare and often very severe disease that occurs when the cell’s energy provider, the mitochondria, do not function properly. Napazimone (KL1333) is in a pivotal clinical study (FALCON), with a positive interim analysis achieved, in adult patients with genetically confirmed primary mitochondrial disease.
READ MORE ABOUT NAPAZIMONE (KL1333) FOR mtDNA PRIMARY MITOCHONDRIAL DISEASE